COVID-19 パンデミックが始まってから数カ月後の2020年初頭、科学者らはCOVID-19感染症の原因ウイルスであるSARS-CoV-2の全ゲノム配列を決定することができた。その時点で、その遺伝子の多くはすでに判明していたが、タンパク質をコードする遺伝子の全容は解明されていなかった。今回、MITの研究者らが広範な比較ゲノム研究を行った結果、SARS-CoV-2のゲノムについて、最も正確で完全な遺伝子アノテーションを作成した。
この研究結果は、2021年5月11日にNature Communications誌のオンライン版に掲載されたが、その中でこの科学者らは、いくつかのタンパク質をコードする遺伝子を確認するとともに、これまで遺伝子として示唆されていたいくつかの遺伝子が、いかなるタンパク質もコードしていないことを発見した。本研究の上席著者であり、マサチューセッツ工科大学コンピュータ科学・人工知能研究所(CSAIL)のコンピュータ科学教授、およびマサチューセッツ工科大学とハーバード大学のブロード研究所のメンバーであるマノリス・ケリス博士は、「我々は、この強力な比較ゲノミクス手法を進化のシグネチャーに用いることで、この非常に重要なゲノムの真の機能的なタンパク質コードを発見できた」と述べている。
また、研究チームは、SARS-CoV-2がヒトに感染し始めてから、異なる分離株で生じた約2,000の変異を分析し、これらの変異が、ウイルスが免疫系を回避したり、感染力を強めたりする能力を変化させる上で、どの程度重要であるかを評価した。このNature Communications誌に掲載されたオープンアクセス論文は、「SARS-CoV-2ゲノムにおけるオーバーラップするORFの矛盾した曖昧な名称。ホモロジーに基づく解決法(Conflicting and Ambiguous Name of Overlapping ORFs in the SARS-CoV-2 Genome: A Homology-Based Resolution)」と題されている。
SARS-CoV-2のゲノムは、約30,000個のRNA塩基から構成されている。科学者らは、関連するウイルスに見られるタンパク質をコードする遺伝子との類似性に基づいて、タンパク質がコードされていることが判っている幾つかの領域を特定した。また、タンパク質をコードしていると疑われる領域も幾つかあったが、これらはタンパク質をコードする遺伝子として明確に分類されていなかった。
SARS-CoV-2のゲノムのどの部分に実際に遺伝子が含まれているのかを明らかにするために、研究者らは、類似したウイルスのゲノムを比較する「比較ゲノミクス」と呼ばれる研究を行った。SARS-CoV-2ウイルスは、サルベコウイルスと呼ばれるウイルスの亜属に属しており、そのほとんどがコウモリに感染する。研究者らは、SARS-CoV-2、2003年のSARS発生の原因となったSARS-CoV、およびコウモリ由来のサルベコウイルス42株について解析を行った。
ケリス博士はこれまでに、この種の解析を行うための計算機技術を開発し、ヒトゲノムと他の哺乳類のゲノムの比較にも利用してきた。この技術は、特定のDNAやRNAの塩基が種間で保存されているかどうかを分析し、その進化のパターンを比較することに基づいている。
これらの技術を用いて、SARS-CoV-2のゲノムには、コロナウイルスでよく知られている5つの遺伝子に加えて、6つのタンパク質をコードする遺伝子が確認された。また、ORF3aと呼ばれる遺伝子をコードしている領域に、さらにORF3cと呼ばれる遺伝子がコードされていることも判明した。この遺伝子は、ORF3aと重複するRNA塩基を持つが、異なるリーディングフレームに存在する。このような「遺伝子の中の遺伝子」は、大規模なゲノムではまれだが、多くのウイルスではよく見られる現象であり、ウイルスのゲノムはコンパクトになるように選択圧がかかっている。この新しい遺伝子の役割は、他のいくつかのSARS-CoV-2遺伝子と同様に、まだ明らかになっていない。
また、これまで可能性のある遺伝子として提案されていた他の5つの領域が機能的なタンパク質をコードしていないことを示し、まだ発見されていない保存されたタンパク質をコードする遺伝子が他にも存在する可能性を排除したのだ。
本研究の筆頭著者でCSAILの研究員であるIrwin Jungreis氏は、「我々はゲノム全体を解析し、他に保存されたタンパク質をコードする遺伝子がないことを確信した」と語る。「未解明の遺伝子の機能を解明するためには、実験的な研究が必要だ。どの遺伝子が実在するのかを決定することで、他の研究者が、タンパク質に翻訳されもしないものに時間を費やすのではなく、それらの遺伝子に注目することができるようになるだろう。」
また、過去の多くの論文では、誤った遺伝子セットが使用されているだけでなく、遺伝子名が矛盾している場合があることも認識した。この状況を改善するために、MITの研究チームは、SARS-CoV-2のコミュニティを集め、SARS-CoV-2の遺伝子の命名に関する一連の推奨事項を、数週間前に『Virology』に掲載された別の論文の中で発表した。その論文は「Conflicting and Ambiguous Names Of Overlapping ORFs in the SARS-CoV-2 Genome: A Homology-Based Resolution」と題されている。
急速な進化
今回の研究では、SARS-CoV-2が最初に発見されて以来、SARS-CoV-2に生じた1,800以上の突然変異を分析した。それぞれの遺伝子について、その遺伝子が過去にどれだけ急速に進化したのかと、今回のパンデミックが始まってからどれだけ進化したのかを比較した。
その結果、ほとんどの場合、パンデミック以前に急速に進化した遺伝子はその傾向が続き、ゆっくりと進化する傾向にあった遺伝子はその傾向を維持していることがわかった。しかし、これらのパターンには例外も見られ、ウイルスがヒトという新しい宿主に適応するためにどのように進化してきたかを解明できるかもしれないと、博士は言う。
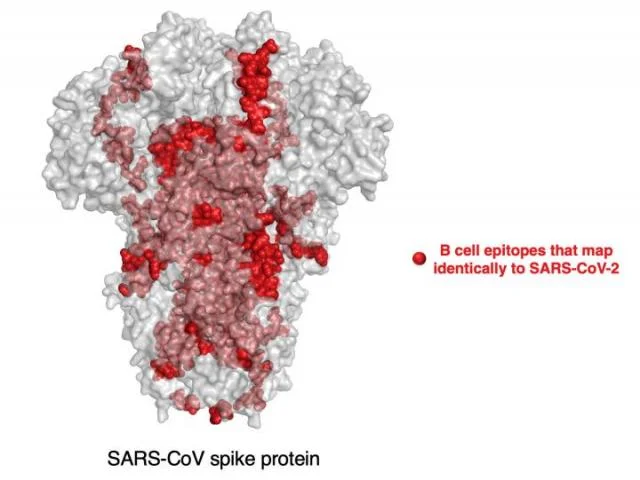
その一例として、ウイルスの遺伝物質を取り囲むヌクレオカプシドタンパク質の領域に、過去の進化パターンから予想されるよりも多くの変異があることが判明した。このタンパク質領域は、ヒトB細胞の標的としても分類されている。したがって、この領域の変異は、ウイルスがヒトの免疫系を回避するのに役立っている可能性があると、ケリス博士は言う。
「SARS-CoV-2のゲノム全体の中で最も加速された領域は、このヌクレオカプシドタンパク質のちょうど真ん中に位置している」と彼は言う。「我々は、この領域に変異を起こさない変異体は、ヒトの免疫系に認識されて排除されるのに対し、この領域にランダムに変異を蓄積する変異体は、実際にはヒトの免疫系から逃れて流行し続けることができると推測している」。
研究者らは、イギリスのB.1.1.7株、ブラジルのP.1株、南アフリカのB.1.351株など、懸念されている亜種に生じた変異も分析した。これらの亜種をより危険なものにしている変異の多くはスパイクタンパクに見られ、ウイルスがより速く広がり、免疫システムを回避するのに役立っている。しかし、これらの変異体は、それぞれ他の変異も持っている。
「これらの変異体には、それぞれ20以上の他の変異があり、それらのうちどれが何かをしている可能性が高く、どれがそうでないかを知ることが重要だ」とJungreis氏は言う。「そこで、比較ゲノミクスの手法を用いて、保存されている位置にあるものから、どれが重要であるかを最初に推測した」。
このデータは、他の研究者が、ウイルスの感染力に大きな影響を与える可能性が高いと思われる突然変異に注目する際に役立つだろう、とこの研究者らは述べている。研究チームは、注釈付きの遺伝子セットとその変異の分類を、カリフォルニア大学サンタクルーズ校のゲノムブラウザで公開しており、他の研究者が利用できるようにしている。
ケリス博士は、「今では、これらの変異体の進化的背景を実際に研究し、現在のパンデミックがその大きな歴史の中でどのように位置づけられるかを理解することができる。多くの変異を持つ株について、どの変異が宿主特異的な適応である可能性が高いのか、どの変異が特筆すべきことではないのかを知ることができる」。
この研究は、米国国立ヒトゲノム研究所および米国国立衛生研究所から資金提供を受けている。また、フラットアイアン研究所計算生物学センターの研究員であるレイチェル・サルフォン博士も、この論文の著者の一人だ。
この記事は、MITのサイエンスライターであるAnne Trafton氏が執筆したニュースリリースに基づき作成されました。