私たちの細胞には、周囲の状況を察知して反応を切り替える「スイッチ」が無数に存在します。しかし、これまでの薬はそのスイッチを「入れるか切るか」という極端な操作しかできず、それが予期せぬ副作用の原因となることもありました。ミネソタ大学医学部を中心とする研究チームは、細胞内で最も重要な役割を果たす受容体を、驚くほど精密に制御する新しい手法を開発しました。分子の「バンパー(緩衝材)」や「グルー(接着剤)」として機能する化合物を用いることで、細胞内のシグナル伝達を「リワイヤリング(配線の書き換え)」し、副作用を抑えたスマートな次世代医薬品への道を開いたのです。

この画期的な研究成果は、2025年10月22日付の学術誌『Nature』に掲載されました。オープンアクセス記事のタイトルは、「Designing Allosteric Modulators to Change GPCR G Protein Subtype Selectivity(GPCRのGタンパク質サブタイプ選択性を変化させるアロステリック調節因子の設計)」です。
最も重要な創薬ターゲット「GPCR」の課題
現在、米国食品医薬品局(FDA)が承認している全薬剤の約3分の1が、Gタンパク質共役受容体(GPCR: G protein-coupled receptor)ファミリーを標的としています。GPCRは創薬において最も成功しているターゲットですが、科学者たちはまだその潜在能力を十分に引き出せていないと考えていました。
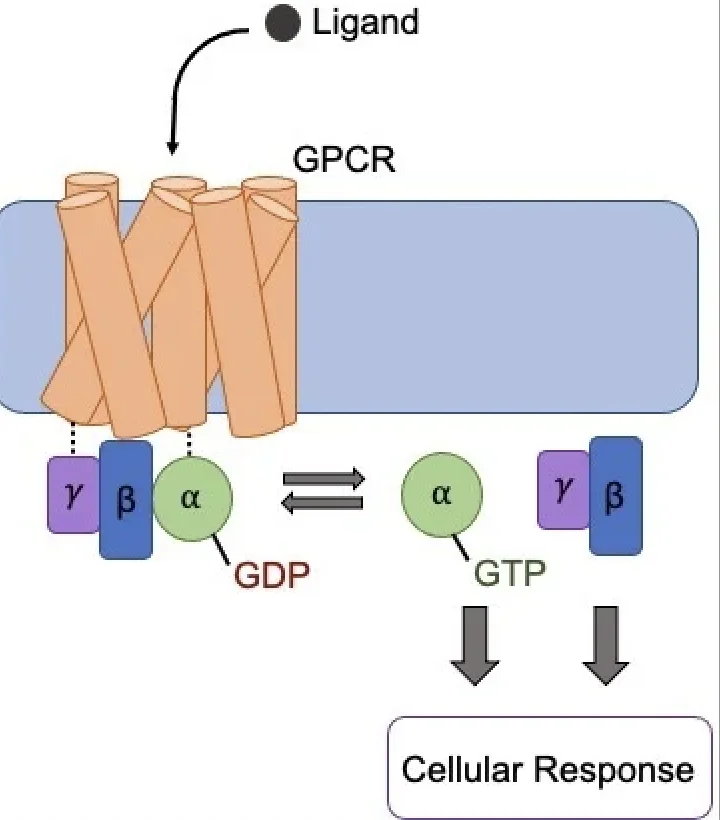
これらの受容体は、16種類の異なるGタンパク質を介して、細胞内で膨大な数のシグナル伝達経路を活性化します。その結果、さまざまな細胞・生理学的効果が生まれますが、中には治療に役立つ経路もあれば、望ましくない副作用を引き起こす経路もあります。これが、新しい治療薬開発の大きな障壁となっていました。
ミネソタ大学医学部助教授で、本研究のシニアオーサーおよび責任著者であるローレン・スロスキー博士(Lauren Slosky, PhD)は次のように述べています。
「特定のシグナルだけを発生させる薬を設計できれば、より安全で効果的な薬が作れるはずです。しかし、これまでその具体的な方法は明らかではありませんでした」
細胞の「内側」から攻める新戦略
カリフォルニア州のサンフォード・バーナム・プレビィス医学研究所(SBP: Sanford Burnham Prebys Medical Discovery Institute)の化学者らを含む研究チームは、受容体が持つ本来のシグナル伝達経路の一部だけを選択的に活性化する化合物の設計戦略を考案しました。
従来のGPCRを標的とした薬のほとんどは、細胞の「外側」から受容体に結合します。しかし、今回の新しい化合物は、これまで未開拓だった細胞の「内側」にある部位に結合します。これにより、受容体がシグナルを伝える相手(シグナル伝達パートナー)と直接やり取りすることが可能になりました。
研究チームがGPCRの一種である「ニューロテンシン受容体1」をモデルに調査したところ、細胞内部の結合部位に作用する化合物が、特定のパートナーとの結合を促進する「分子グルー(接着剤)」や、逆に結合を阻害する「分子バンパー(緩衝材)」として機能することがわかったのです。
ローレン・スロスキー博士は、「ほとんどの薬は受容体のすべてのシグナルを一律に『上げる』か『下げる』かすることしかできません。しかし、今回の新しい化合物は単なる『音量調節』ではなく、細胞が受け取る『メッセージの内容』そのものを変えることができるのです」と説明します。
科学的に「予測可能」な創薬へ
科学者たちはモデリング技術を駆使して、多様なシグナルプロファイルを持ち、異なる生物学的効果をもたらす新しい化合物を設計しました。
研究の共同著者であり、SBPの創薬化学エグゼクティブ・ディレクターを務めるスティーブン・オルソン博士(Steven Olson, PhD)は次のように語っています。
「化合物の化学構造を変えることで、どのシグナル経路をオンにし、どれをオフにするかを自在にコントロールできました。最も重要なのは、これらの変化が予測可能であり、創薬化学者が合理的に新薬を設計するために活用できるという点です」
ニューロテンシン受容体1における最終的な目標は、副作用を最小限に抑えた慢性疼痛や依存症の治療薬を見つけることです。この細胞内結合部位はGPCRスーパーファミリーに共通するものであるため、この戦略は多くの受容体に応用できる可能性が高く、幅広い疾患に対する革新的な治療法につながることが期待されています。